Please wait for operation to finish...
Please wait while GAM is initializing...
Your session has ended. If you wish to restart, please reload the page.
Shiny GAM: integrated analysis of genes and metabolites
Shiny GAM is deprecated in favor of Shiny GATOM, next generation of the metabolic network analysis pipeline

Help
This is GAM (“Genes And Metabolites”): a web-service for integrated network analysis of transcriptional and steady-state metabolomic data focused on identification of differential metabolic subnetworks most changing between two conditions of interest.
The analysis consists of two steps:
- Step 1: Creating a network of metabolites and reactions specific to the provided data.
- Step 2: Finding a connected reaction module that contains the most significant changes.
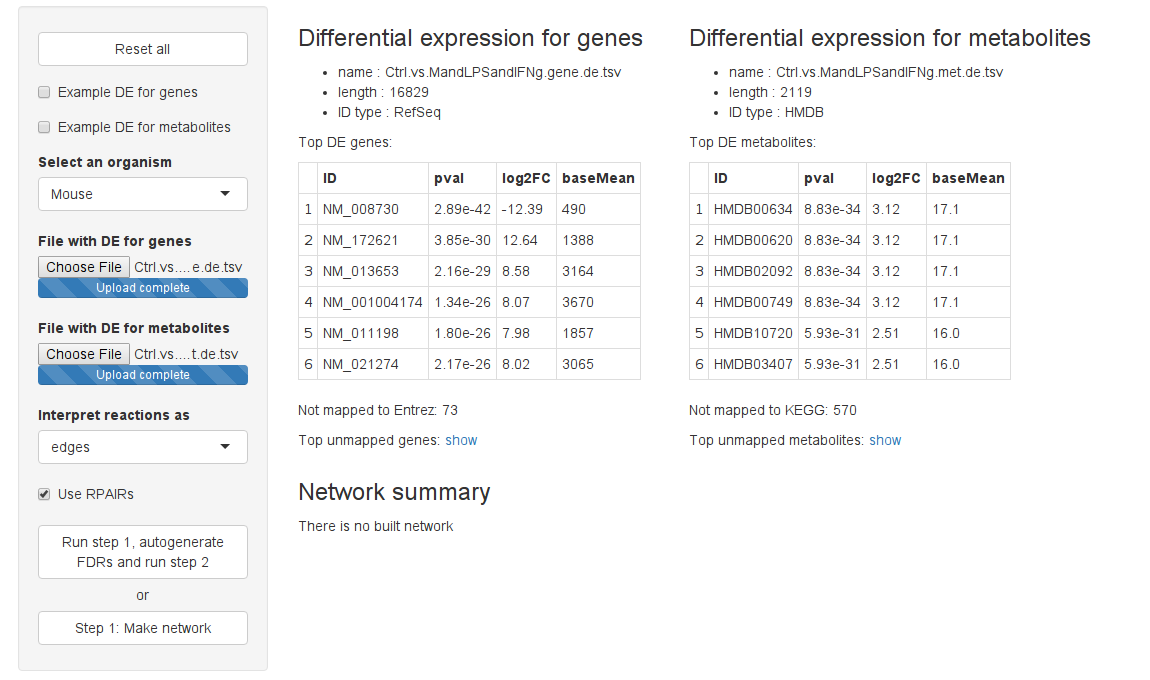
Reset all button allows to start the analysis from the beginning, clearing the input data sets. Example DE for genes and Example DE for metabolites checkboxe automatically upload corresponding example data for mouse macrophages M1 vs M2 activation.
Constructing a network
To construct a network select an organism from the Select an organism dropdown menu. Only reactions possible in the selected organism are used. Currently supported organisms are Homo sapiens, Mus musculus, Arabidopsis thaliana and Saccharomyces cerevisiae. Please, send requests for additional organisms to Alexey Sergushichev at asergushichev @ path.wustl.edu.
The next step is to upload differential expression (DE) data for genes and/or metabolites. GAM can be run using either gene DE data or metabolite DE data or both datasets. Each DE dataset must be in a separate CSV file (comma-, tab- and space- separated files are supported, archived files are supported too). The first line of each file must contain a header with the column names. Files should contain the following columns:
- “ID”: RefSeq mRNA transcript ID, Entrez ID or symbol for genes and HMDB or KEGG ID for metbolites.
- “pval”: Differential expression p-value (non-adjusted).
- “log2FC”: Base 2 logarithm of the fold-change.
NB: Columns in your files can have somewhat different names and GAM will try to guess which one to use. If you favorite DE tool produce names that GAM can not recognize, please, contact Alexey Sergushichev.
The “log2FC” column is optional, but we recommend to provide it if possible. It is used for colors in a graph visualization. Any other columns will be copied to a network as node or edge attributes. Example data can be downloaded:
- for genes and
- for metabolites.
We also provide example data with differential gene expression for
- human MCF10A cell line treated with 2DG (from GSE59228),
- arabidopsis wild-type vs allene oxide synthase (AOS) knock-out mutant (from Landesfeind et al, PeerJ 2014),
- yeast response to hypoxia (from GSE9514).
After files are uploaded, a file summary is displayed. In the bottom of the summary rows with IDs that were not mapped to the IDs used in network (e.g. Entrez for mouse) are shown. Verify that the files were parsed correctly.
If present, DE for genes is converted to DE for reactions. This is done by considering for a reaction all the genes that code any enzyme that takes part in this reaction. Gene with the minimal p-value is selected and its p-value is assigned as the reaction p-value. All reactions without p-values are discarded as having no expressed enzymes. We recommend to exclude genes with low expression prior uploading.
Reactions in the network can be interpreted as either edges or nodes. Select one of the interpretations. See the image below.
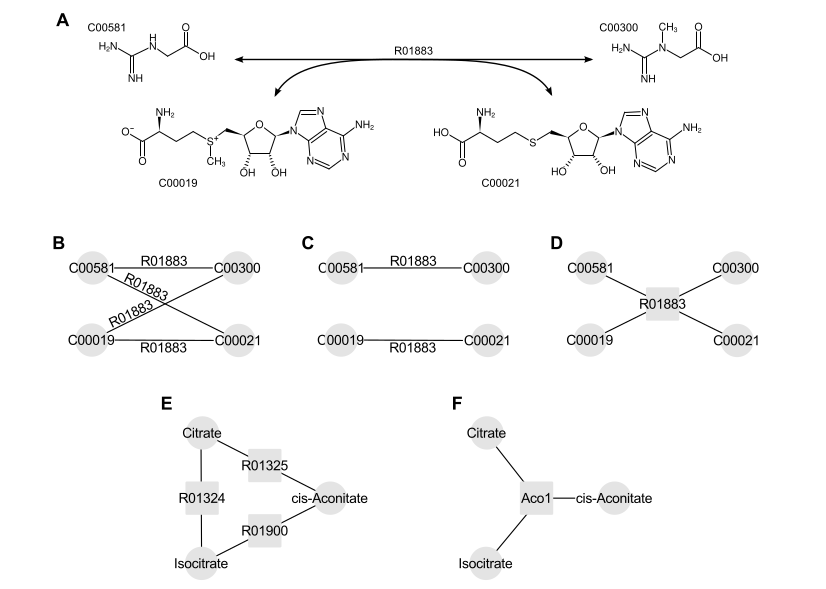
Reactions as edges. If reactions are interpreted as edges, if a pair of metabolites has multiple reaction connections between them, only the reaction with the minimal p-value is kept. All the remaining reactions and metabolites that take part in these reactions are combined into a network as edges and nodes respectively (see panel B on the image above). To only consider substrate-product pairs that make up a main type KEGG reaction pair (RPAIR), as opposed to all substrate-product pairs, select the Use RPAIRs option (panel C).
Reactions as nodes. If reactions are interpreted as nodes, then both metabolites and reactions are added as nodes to the network. Edges are added between a metabolite and a reaction if the metabolite takes part in this reaction (panel D). We recommend to collapse groups of reactions that have at least one common metabolite and the same most significant gene into single nodes with Collapse reactions option (panels E and F). It removes artificial biases created by clusters of similar reactions.
Generally, all of these options will lead to similar results. We recommend to use the automatically selected option values as this makes the network simpler and the analysis faster.
Click Step 1: Make network to finish this step.
Finding a module
After making a network, you can find a connected module that contains the most significantly changed genes and metabolites. Internally, this is done by first scoring nodes and edges based on their p-values in such way that positive scores correspond to significant p-values and negative scores correspond to insignificant changes. Then the problem of finding a connected subgraph with maximum summary weight (maximum-weight connected subgraph, MWCS problem) is solved.
The FDR values and Score for absent metabolites options control the size of the module. Increasing/decreasing the FDR for reactions (FDR for metabolites) value makes adding reactions (metabolites) to a module easier/harder. We recommend to start from the default values and then gradually change them depending on the results. Default FDR values are generated so that the module will a size of ~100 reactions (Autogenerate FDRs button).
There are three solvers available to solve MWCS instance: heinz, heinz 2 and gmwcs. Heinz solver (Dittrich at al. 2008) is called when the option Try to solve to optimality selected. Solving to optimality can take a relatively long time, so we provide two other solvers that can be run for a predefined amount of time. These are Heinz 2 (El-Kebir&Klau 2014, https://software.cwi.nl/cwisoftware/software/heinz), which is used when edges are not scored, and gmwcs (https://github.com/ctlab/gmwcs-solver, http://arxiv.org/abs/1605.02168), which is used when edges are scored.
Click Step 2: Find module button to find a module in the network. The module will be shown on the right panel.
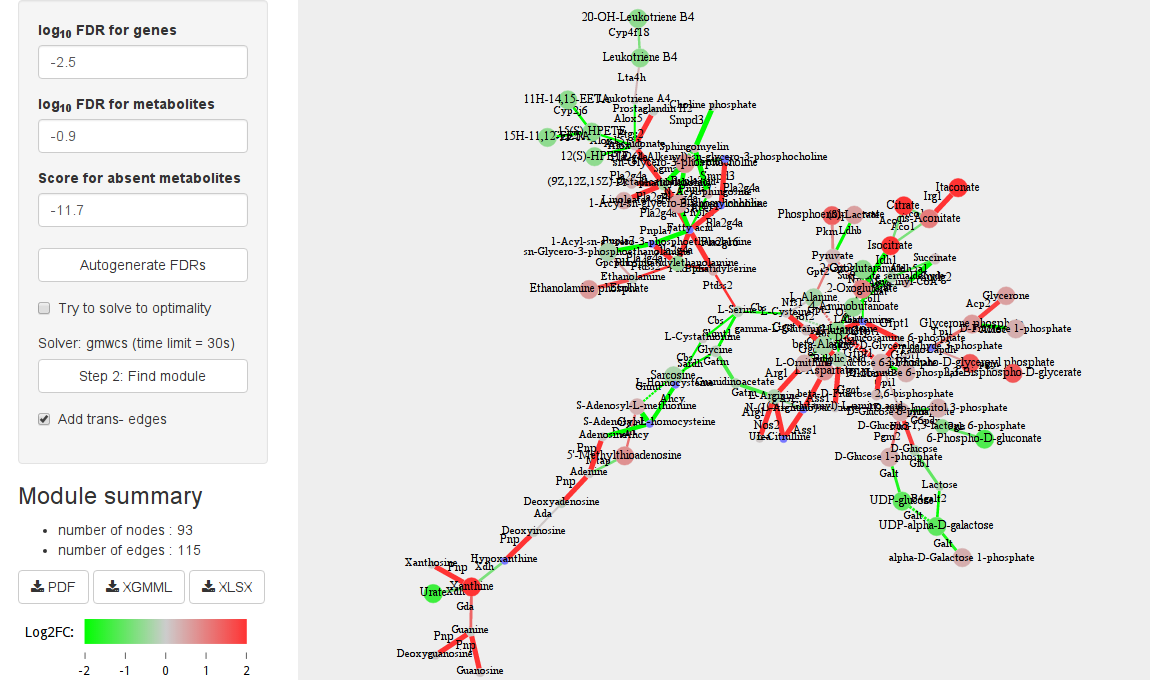
Graph legend
We use the following scheme:
- Red nodes and edges are up-regulated (log2FC > 0).
- Green nodes and edges are down-regulated (log2FC < 0).
- Blue nodes and edges don't have log2FC values.
- Bigger size of nodes and width of edges means lower p-values.
- Dashed edges are trans-RPAIRs.
- Circle nodes represent metabolites, square nodes represent reactions.
Additionally, raw field values are displayed in the node and edge tooltips.
Post-processing
There are couple of post-processing steps that are available. If in the network
reactions are edges and RPAIRs are used, then trans-connections can be added.
When reactions are nodes, metabolites that take part in at least two reactions
from the module can be added into the module. Both this steps add elements
that are not required for connectivity, but could help to interpret the results.
Saving the module
You can download the module in several formats by clicking corresponding links:
- PDF file contains the image of the module, as it appears on the web-site,
- XGMML file contains module that can be imported into Cytoscape (see Importing to Cytoscape section below),
- XLSX file contains simple table of metabolites and reactions in the module, along with corresponding fields (logPval, log2FC, …). button.
Interpreting the results
The result of the analysis is a set of connected most regulated reactions. These reactions are associated with strongly differentially changing enzymes and metabolites or are closely connected to such reactions which implies regulated flux. Inside the module one would typically consider small groups of particularly significant reactions or individual reactions close to the center of the module. Such groups or individual reactions are implied to be of biological importance for the considered process. These can lead to potential experimental designs including labeling, media perturbation or gene knockout/knockdown experiments. For examples see Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization and Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth papers.
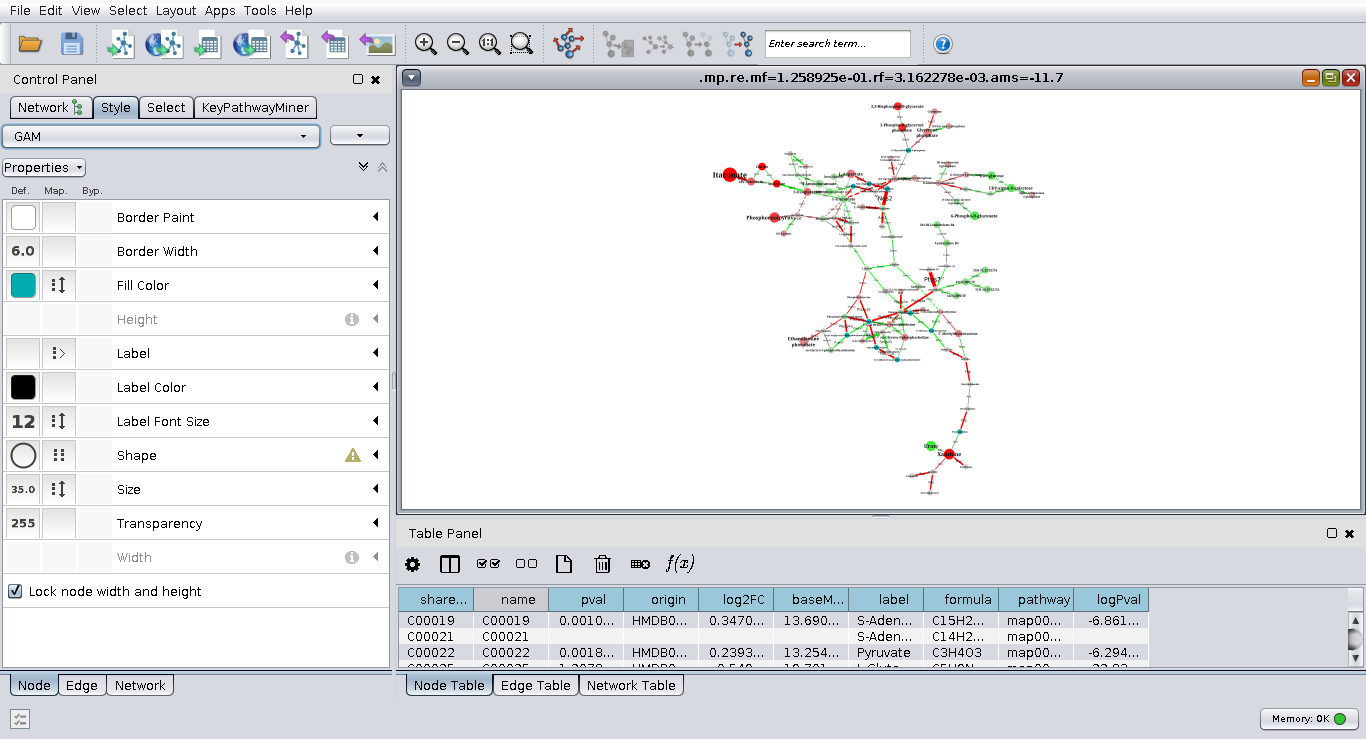
Importing module into Cytoscape
XGMML file with module can be imported into Cytoscape for consecutive exploring and processing. For the best experience we recommend to use GAM's VizMap style for Cytoscape that can be downloaded here.
You can load XGMML file (e.g. one generated for default parameters) using menu File>Import>Network>File… and selecting the file. Press OK on the pop-up dialog with import options. The module will appear as a single node, use menu Layout>Apply preferred layout to do a layout. Then load GAM Cytoscape VizMap style using menu File>Import>Style… and then selecting it in the Style tab on the right, instead of default. The result should look like this:
About
By Alexey Sergushichev alserg@corp.ifmo.ru and co
You can find source code of this site here.
Citation: Sergushichev et al. GAM: a web-service for integrated transcriptional and metabolic network analysis. Nucleic Acids Research. http://dx.doi.org/10.1093/nar/gkw266
Powered by R Shiny.
The authors thank Nicole Rockweiler for the help with improving the web-service.